
In this quick tutorial you'll learn how to draw a Spruce Tree in 5 easy steps - great for kids and novice artists.
The images above represents how your finished drawing is going to look and the steps involved.
Below are the individual steps - you can click on each one for a High Resolution printable PDF version.
At the bottom you can read some interesting facts about the Spruce Tree.
Make sure you also check out any of the hundreds of drawing tutorials grouped by category.
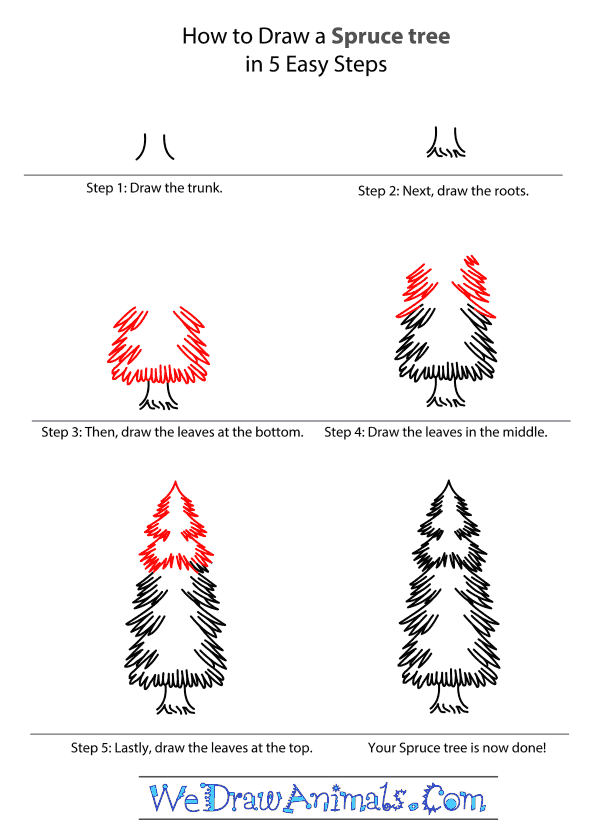
How to Draw a Spruce Tree - Step-by-Step Tutorial

Step 1: To start, draw the trunk. Draw a small curved in line, leave some space and draw another small line that curves in toward the middle.

Step 2: Next, draw the roots. Draw a small “V” under the trunk to the left, leave spaces and draw a few small wavy lines until you get to the right side of the trunk.

Step 3: Now, draw the leaves at the bottom of the tree. Draw a straight across line of curved spikes along the top of the trunk. Make sure it is a lot longer than the trunk opening. Draw a slight slanted inward line of curved spikes going up from both ends of the leaves you just drew. Do not connect them at the top yet.

Step 4: Then, draw the leaves in the middle of the tree. Continue drawing your slight slanted line of curved spikes like you did for the bottom leaves. Make sure to draw the leaves on both sides of the tree.

Step 5: Finish by drawing the leaves at the top. Continue your slight slanted line of curved spikes until it is almost to a point at the top of the tree and then stop. Draw a medium sized triangle with no bottom then connect both sides of leaves.
Interesting Facts about Spruce Trees
Spruce Trees are cone shaped evergreens that grow in high places like mountains. In nature they grow in places like Sweden and Norway. Spruce Trees grow between 66 and 197 feet tall. Their needles grow in a spiral pattern. Every 4 to 10 years the tree sheds its needles and grows new ones. The needs are curved a little and measure about ? of an inch. Spruce Trees also reproduce by growing pinecones. A Spruce’s cone is long and cylinder-like.
Did you know?
- Spruce Trees are common Christmas trees.
- The states Alaska, Colorado, South Dakota, and Utah in the United States as use a type of Spruce Tree for their symbol.
- There are 35 species of Spruce Tree. The most popular are the Blue Spruce, the Red Spruce, and the White Spruce.
- The Wright Brothers built their first flying machine “The Flyer” out of spruce wood.
- In Bavaria the Spruce Tree is a symbol of life. It is often used to make maypoles for celebrations.
Theme: Spruce Trees are the perfect topic for a Christmas time lesson plan. First decide which tree you want to highlight this Christmas. Then set up the evergreen of your choice. Let your students decorate the tree and while you are at it create a science lesson on the Spruce Tree.
How to Draw a Spruce Tree – Step-by-Step Tutorial